

 English
English French
French Spanish
Spanish Russian
Russian Korean
Korean Japanese
JapaneseStudy on Different Types of Ginseng Extract Ginsenoside
Ginsenosides (GS) are the main active ingredients in precious medicinal herbs such as ginseng, panax notoginseng and American ginseng. They belong to the triterpenoid glycoside class and are composed of a glycoside (ginsenoside) and a sugar group [1]. Ginsenosides have a wide range of biological activities, such as anti-tumor [2], anti-inflammatory [3], anti-fatigue [4-5] and anti-oxidation [6], etc., and secondary ginsenosides exhibit even more excellent activities. However, the highly active secondary ginsenosides have problems such as low content, poor water solubility, low bioavailability and short half-life [7], which limits the application of ginsenosides in the fields of food health care and biomedicine.
Structural modification is an important means to improve the biological activity of ginsenosides, improve pharmacokinetic properties and reduce toxicity. Ginsenosides are usually composed of a hydrophobic aglycone linked to 1–4 hydrophilic sugar moieties, so they can be modified in these two ways. At present, the modification of ginsenosides mainly adopts chemical modification strategies, and ginsenoside derivatives with better activity and physicochemical properties are obtained by modifying the aglycone structure or modifying the sugar chain [8]. In terms of structure, the cycloalkane structure of the aglycone is stable, and it is difficult to directly modify the aglycone backbone.
The current main modification strategies focus on the hydroxyl group on the aglycone, and derivatives with diverse structures are obtained through synthetic methods such as esterification, oxidation, the introduction of heterocycles, or molecular hybridization. Modifications to the sugar chain mainly involve the extension of the sugar moiety or modification of the hydroxyl group on the sugar chain. Studies have shown that the type, number and binding site of the sugar group on the ginsenoside parent nucleus are closely related to the biological activity of ginsenosides [18-20]. In general, the relationship between the number of sugar groups and the anti-tumor activity of ginsenosides is as follows: aglycone > monosaccharide glycoside > disaccharide glycoside > trisaccharide glycoside > tetrasaccharide glycoside. Therefore, modifying the sugar chains of ginsenosides is of great significance for improving their biological activity. This paper reviews recent progress in the chemical modification and biological activity of ginsenosides, elucidates the structure-activity relationship, and summarizes the characteristics and laws of structural modification of ginsenosides, providing a reference for subsequent structural modification.

1 Classification and structural characteristics of ginsenosides
According to the different structures of the aglycone, they can be divided into three types: dammarane type, oleanane type and ocotillol type. Dammarane-type ginsenosides can be further subdivided into protopanaxdiol (PPD) and propanaxatriol (PPT) according to the position of the substituent group attached to the aglycone.
1.1 Dammarane type
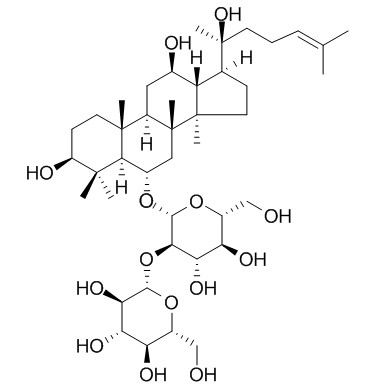
Dammarane-type ginsenosides include PPD and protopanaxatriol (PPT), which are tricyclic triterpene saponins. Common protopanaxadiol ginsenosides include C-K (1), Rh2 (2), Rd (3), Rg3 (4), Rb1 (5), Ra1, Ra2, Ra3, Rb2 and panaxadiol (PD) (Fig. 1). Since ginsenosides 1, 2, 4 and 5 have stronger biological activity, their synthesis and modification have attracted much attention [9-11]. Common protopanaxatriol-type ginsenosides mainly include Rh1 (6), Rg1 (7), Rg2 (8), Re, Rf, F1, F3, F5 and glycosylated panaxatriol (PT) (see Figure 2), among which 6, 7 and 8 have been studied extensively [8, 12].
1.2 Oleanolic acid type
Oleanolic acid type ginsenosides are pentacyclic triterpene saponins, which are formed by the glycosylation of oleanane type saponins (OATS) at the C-3 and C-28 positions. Common oleanolic acid-type ginsenosides include R3 (9), Ro (10), and R4 (11), among others [13-14] (see Figure 3).
1.3 Ocotillol types
Ocotillol typesapogenins (OTS) can form four conformations: (20S, 24S), (20R, 24R), (20S, 24R) and (20R, 24S), depending on the configuration of the C-20 and C-24 linked sugar moieties. Commonly-found ginsenosides of the Oxytropae type include F11 (12), RT5 (13), RT2 (14), etc. [15-17] (see Figure 4).
2 Structural modification of ginsenosides and structure-activity relationships
2.1 Esterification modification
Ginsenosides have poor water solubility and low fat solubility, resulting in low bioavailability and ineffective health maintenance and therapeutic effects. The ideal lipophilicity of a drug molecule needs to be within a certain range to ensure its bioavailability and clinical efficacy [8]. Pharmacokinetic studies have shown that ginsenosides are hydrolyzed by intestinal flora after oral administration, and the metabolites produced by hydrolysis are absorbed into the liver through the vein, where they react with fatty acids to form fatty acid ester compounds [21]. Further studies have found that ginsenoside derivatives linked to fatty acids have low cytotoxicity in cells, a long residence time, and a more lasting effect. On the other hand, because cell membranes are mainly composed of lipids, lipophilic ester derivatives can enhance the oral absorption of undesirable drugs by improving the overall membrane permeability. These findings provide ideas for the modification of ginsenosides. The modification of ginsenosides using acids such as organic acids (fatty acids, aromatic acids, anhydrides), amino acids and inorganic acids (sulfuric acid) is an important strategy for the study of ginsenoside derivatives.
2.1.1 Organic acid modification
Liang et al. reacted the hydroxyl group at the C-3 position of ginsenoside Rh2 (20S-Rh2, 2) with two 6-maleimidocaproic acid and 11-maleimidoundecanoic acid derivatives with hydrophilic functional groups and different carbon chain lengths to obtain the esterified derivatives 15 and 16 (see Figure 5). Compared with Rh2, the solubility of the two modified products increased by about 4 times and 2 times, respectively. In vitro anti-proliferation activity tests showed that compound 15, which has a shorter carbon chain, exhibited higher inhibitory activity against the HeLa cell line, while compound 16, which has a longer carbon chain, did not show anti-proliferative activity [22]. Li et al. reacted decanoic acid, cyclohexanecarboxylic acid and isobutyric acid reacted with the C-20 hydroxyl group of ginsenoside C-K to synthesize three ginsenoside C-K monoesterified derivatives (17–19) [23] (see Figure 5).
In terms of growth inhibition of the breast cancer cell line MCF-7, the inhibitory activity of compounds 18 and 19 at 25 μmol/L was significantly higher than that of ginsenoside C-K, while compound 17 did not show any inhibitory effect, indicating that ginsenoside derivatives modified with short-chain fatty acids have stronger anti-tumor activity than those modified with long-chain fatty acids. Other studies [24–27] have also shown that modified short-chain fatty acid saponin derivatives not only have optimized physicochemical properties, but also have better anti-tumor effects than long-chain fatty acid esters.
Li et al. synthesized a fully acetylated derivative of ginsenoside C-K by polyesterification modification, except for the glucose-based C-6 hydroxyl group [28] (see Figure 5). Anti-tumor activity tests showed that compared with C-K, it can inhibit the proliferation of multiple tumor cell lines at lower concentrations, while significantly inhibiting tumor growth in a hepatocellular carcinoma xenograft model without side effects on major organs.
It can be seen that after esterification modification of ginsenoside C-K, its cytotoxicity is reduced and its antitumor activity is increased. Wang et al. reacted the C-3 hydroxyl group of PD with benzoic acid derivatives, amino acids and tetrachlorophthalic anhydride to obtain a series of PD derivatives 21–31 [29] (see Figure 5). Anti-tumor proliferation tests showed that most of the compounds had inhibitory effects on cancer cell lines, including human liver cancer cells HepG-2, human lung cancer cells A549, human breast cancer cells MCF-7, and human colon cancer cells HCT-116. Compared with PD, ginsenoside derivatives 22, 23, and 26 showed significant inhibitory effects on cancer cell proliferation. for example, 22 had the lowest IC50 value for A549 (IC50 = 18.91 ± 1.03 μmol/L), while for MCF-7 cells, compound 23 showed better inhibitory activity (IC50 = 8.62 ± 0.23 μmol/L). This result shows that the introduction of an aromatic acid into ginsenosides can also significantly improve antitumor activity.
The above-mentioned structure-activity relationship shows that the introduction of short-chain fatty acids into ginsenosides exhibits better activity than long-chain fatty acids. The number of esterification modification sites (monoesters and polyesters) and the type of acid (fatty acids and aromatic acids) have no significant effect on biological activity.
2.1.2 Amino acid modification
25-Hydroxyl-protopanaxdiol (25-OH-PPD) (34), a natural compound isolated from ginseng fruit, has significant antitumor activity and the advantages of low side effects and high absolute oral bioavailability. Yuan et al. [30] combined it with non-protein amino acids that play unique physiological functions and medicinal values, designed and synthesized a series of new 25-OH-PPD derivatives 33–45 (see Figure 6). Antitumor activity showed that some 25-OH-PPD derivatives exhibited excellent inhibitory activity against tumor proliferation. For example, compound 33 showed strong antitumor activity against the HCT116 and BGC-823 cell lines, with IC50 values of 4.76 μmol/L and 6.38 μmol/L, respectively (see Table 1). In addition, amino acid derivatives of 25-OH-PPD (46–59) also exhibited antitumor activity [31] (see Table 1).
As can be seen from Table 1, the IC50 values of some non-protein amino acid modified products are greater than 100 μmol/L, while the anti-tumor activity of protein amino acid modified products is generally better than that of non-protein amino acid modified products, and their IC50 values are all lower than 30 μmol/L. On the other hand, the amino acids on ginsenoside derivatives with an IC50 value of less than 10 μmol/L for anti-tumor proliferation all have Boc protective groups, and removing the Boc protective groups significantly reduces the anti-tumor activity of the product, which indirectly indicates that increasing the lipid solubility of the product through esterification can significantly increase the biological activity of ginsenoside derivatives.
2.1.3 Inorganic acid modification
At present, inorganic acid modification mainly uses the sulfonating reagent chlorosulfonic acid to react with the hydroxyl groups on the ginsenoside sugar chain to form a sulfonate, which is then converted to a salt by neutralization with pyridine. As the introduction of a sulfate group increases the polarity of the ginsenoside derivative, solubility is improved. It has been reported that the anticancer activity of sea cucumber saponins, which have a similar structure to ginsenosides, is related to the sulfate group,the fewer sulfate groups present on the sugar chain, the stronger the anti-cancer activity [32].

Based on these findings, Guo et al. [33] used the chlorosulfonic acid-pyridine method to sulfate modify ginsenosides. The resulting derivative SMTG-d3 enhanced natural killer cell activity by promoting the proliferation of T lymphocytes and the production of IFN-γ and TNF-α cytokines. Compared with ginsenoside, SMTG-d3 not only reduces cytotoxicity, but also further enhances antitumor immune activity. Previously, Fu et al. [34-36] also used this method to convert the C-6 hydroxyl group of 20(S)-ginsenoside Rh2 to a sulfonate ester and synthesized two new derivatives 60 and 61 with greatly improved solubility (see Figure 7). Further studies have found that both derivatives can enhance anti-inflammatory and immune effects by blocking mitogen-activated protein kinase and the release of pro-inflammatory mediators induced by activation. This shows that the sulfation of ginsenoside derivatives can increase their solubility, thereby enhancing activities such as anti-inflammatory and anti-tumor effects.
2.2 Oxidative modification
The planar double bond on the ginsenoside side chain and the hydroxyl group in the aglycone structure provide reaction sites for oxidation modification, making it possible to oxidize the C-17 side chain and A and C rings of some ginsenosides. Studies have shown that the double bond on the ginsenoside side chain is one of the reasons for its low solubility [37]. Ginsenosides can increase their solubility in water by reducing the degree of unsaturation or adding ionizable groups such as carboxyl groups through oxidative modification, thereby enhancing their biological activity.
Wong et al. [38] oxidized the double bond on the side chain of ginsenoside 20(R)-Rh2 (2) to obtained a derivative 20(R)-Rh2E2 (62) that can effectively prevent the development of colorectal cancer induced by oxidized azomethane/dextran sulfate sodium salt (AOM/DSS) (see Figure 8). This epoxide compound also has inhibitory activity against other cancer cell lines. For example, its IC50 for lung cancer cells (LLC-1) is 56 μmol/L.
It has been found that PPD is metabolized in the human liver to form C-20-24 epoxide, which contains the Pyxinol skeleton and has good anti-inflammatory activity [39]. Wang et al. epoxidized 20(S)-PPD and then subjected to Dess-Martin oxidation, selective reduction with NaBH4, condensation and deprotection reactions to obtain a series of amino acid-modified C-12 oxidized Pyxinol derivatives [40] (see Figure 9). In vitro cytotoxicity tests showed that most derivatives did not exhibit significant toxic effects.
Using the Griess method to test the inhibitory activity of these derivatives on nitric oxide in RAW264.7 macrophages, derivatives 63a, 63b, 63c, 63d, 64e, 66b, and 66c showed good anti-inflammatory activity (inhibition rates of 48% to 85%), even better than Y13 (known as the Pyxinol derivative with the best anti-inflammatory activity at the C-12 site, with a hydroxyl group, and an inhibition rate of <40%) and the clinically approved glucocorticoid steroid drug hydrocortisone sodium succinate. A structure-activity relationship study showed that oxidation of Pyxinol at the C-12 position can effectively improve the anti-inflammatory activity of derivatives modified at the C-3 position. In particular, N-Boc-protected aromatic amino acids can significantly enhance their anti-inflammatory activity. At the same time, derivatives with the absolute configuration of R at the C-24 position are more active.
Wang et al. [41] used a similar method to selectively oxidize the C-3 position of the Pyxinol skeleton A ring and simultaneously introduce a Michael acceptor to prepare 24 novel ginsenoside derivatives (67a-67h, 68a-68h, 69a-69h) (see Figure 10). The structure-activity relationship shows that the fusion of ginsenoside PPD with a Michael acceptor can enhance the anti-inflammatory activity of the derivative, and the presence of an electron-withdrawing group on the Michael acceptor further enhances the anti-inflammatory activity. The anti-inflammatory activity of the derivative obtained by modifying the C-20 position of PPD with a tetrahydrofuran ring was greatly reduced, but the anti-inflammatory activity of the derivative in which the A ring was oxidized was almost unaffected, which further indicates that the anti-inflammatory biological activity of some ginsenosides can be significantly improved by oxidation modification.
Zhang et al. [42] hydrolyzed PD and oxidized it using pyridinechlorochromate (PCC), O2, and H2O2 to obtain a series of C-17 side chain and A and C ring oxidation derivatives (see Figure 11). Antitumor cell tests showed that some compounds exhibited better antiproliferative activity than the positive control in six cell lines, including A549 (human lung cancer), 8901 (human ovarian cancer), and other cell lines. For example, in the U87 (human glioma) cell line, compounds 70, 78, 82 and 83 were more effective than the positive control, with compound 82 having an IC50 of 19.51±1.00 μmol/L. In the MCF-7 (human breast cancer) cell line, compared with 5-fluorouracil and PD, compounds 71 and 82 exhibited better antitumor activity (IC50 = 17.73~23.58 μmol/L); compounds 71 and 74 also exhibited good antiproliferative activity in HeLa cells. Studies have shown that introducing an enol structure at the α-site of the A ring of PD derivatives can improve their antitumor activity, but not all oxidative modifications of PD derivatives achieve this effect. For example, the antiproliferative activity of compound 81, which was obtained by further oxidation with H2O2, was reduced.
2.3 Heterocyclic modification
Heterocyclic compounds are often used in drug design and synthesis because of their structural diversity and wide range of biological activities, which provides an expansion of the available space for drug-like chemistry. Most marketed drugs contain heterocyclic structures, with nitrogen heterocycles being the most common in marketed drug structures. The nitrogen heterocycles and oxygen heterocycles commonly found in drug molecules contain lone pairs of electrons, which can form hydrogen bonds, which is conducive to improving water solubility and thus bioavailability. Piperazine rings and piperidine rings are very common nitrogen heterocyclic structures in marketed drugs. They can be further derivatized to establish a small compound library [43], which is conducive to designing more compounds for in-depth structure-activity relationship studies. Studies have shown that the introduction of heterocycles in natural products can greatly enhance the biological activity and solubility of derivatives through the principle of “pharmacophore combination” and an increase in the number of hydrogen bonds [44-45]. At present, a large number of studies have reported on the modification of ginsenoside derivatives with heterocycles. Among them, nitrogen-containing heterocyclic compounds have low cytotoxicity, and exhibit good water solubility, permeability and bioavailability.
Pyrazoles are five-membered heterocyclic compounds composed of two adjacent nitrogen atoms. They have a variety of pharmacological activities such as anti-inflammatory, antiviral and antidepressant effects, and are widely used in new drug development [46]. Isoxazole derivatives also have a variety of biological activities such as antibacterial, antiviral and antitumor effects, and are widely used in organic synthesis [47]. Based on this, Dai et al. [48] introduced the pyrazole and isoxazole skeletons into the C-3 position of PD, designed and synthesized 19 PD derivatives containing heterocycles (see Figure 12), and studied their antiproliferative activities against four different tumor cells. The results showed that the products 86 and 87 obtained by fusing the A ring of PD with the pyrazole ring have significant anti-cancer activity. For example, 86 has an IC50 of 14.15±1.13 μmol/L against HepG-2 cells, and 87 has an IC50 of 13.44±1.23 μmol/L against A549, which is four times that of PD. It also has a greater inhibitory effect on the other three tumor cells. However, compounds 88 and 89a–89i have poor water solubility due to the presence of multiple ester bonds and hydrophobic groups such as aromatic rings, resulting in poor anti-tumor proliferation activity. Substituting the ester bonds in the structures of derivatives 88 and 89a–89i with amide bonds to obtain derivatives 90a–90f did not improve the anti-tumor proliferation activity. On the other hand, the results of in vitro activity tests showed that 87 > 86, 89a > 88, and 90a > 91, which indicates that pyrazole-modified PD derivatives are generally more active than isoxazole compounds.
Pyrazine and pyrimidine compounds exhibit a wide range of biological activities, and these two types of structures are often found in marketed drug molecules [49-50]. Wang et al. [51] introduced heterocycles such as pyrazine, oxadiazole, isoxazole, pyrazole and pyrimidine heterocycles were introduced into the C-2 and C-3 positions of PPD via classical organic reactions such as oxidation, hydrogenation, Claisen ester condensation, reduction, and the protection and deprotection of hydroxyl groups. A series of heterocyclic fused 20(S)-PPD derivatives were synthesized (see Figure 13) and their inhibitory effects on receptor activator for nuclear factor-κ B ligand (RANKL)-induced osteoclast differentiation were evaluated. The structure-activity relationship shows that compared with PPD, in addition to the phenylpyrazole derivatives, the inhibition of osteoclast differentiation by the oxadiazole, isoxazole and pyrazole derivatives with five-membered heterocyclic modifications (93, 94 and 95a) have similar or slightly stronger inhibitory activity (IC50= 10.3 μmol/L) than PPD; while the inhibitory activity of compounds modified with six-membered heterocyclic rings such as pyrazine and pyrimidine (92, 96a) is significantly increased.
Based on the excellent activity of compound 96a, the research group further modified the pyrimidine derivatives (see Figure 14). The results showed that at a moderate concentration of 1.0 μmol/L, most derivatives had almost 100% inhibitory effect (except 96f); at a concentration of 0.1 μmol/L, the inhibition effect of the methyl (96b) and ethyl (96c) modified derivatives was significantly enhanced, while the inhibition activity of the methoxy (96d), ethoxy (96e) and amino (96g) modified compounds remained almost unchanged. The researchers further structurally modified 96b with a C-12-hydroxy or C-17 side chain (see Figure 14). The results showed that replacing the hydroxyl group at the C-12 position with a ketone (98), oxime (99), α-hydroxy (100) or acetate (101) group resulted in a significant decrease in inhibitory activity. At a concentration of 0.01 μmol/L, 98–101 showed almost no inhibitory effect. Compound 105 showed the best inhibitory activity (IC50 = 11.8 nmol/L), even at a concentration of 0.01 μmol/L, which was better than PPD activity (IC50 = 10.3 μmol/L), and it could inhibit osteoclastogenesis both in vitro and in vivo.
2.4 Polymer modification
Hydrophilic polymer-modified anticancer drugs can not only compensate for their poor targeting, but also improve the water solubility, stability, in vivo half-life and bioavailability of the drug [52]. In recent years, drug delivery technology has developed rapidly, making ginsenosides widely studied. Lu et al. [53] prepared Rh2 conjugates with water-soluble O-carboxymethyl chitosan (O-CMC) (Rh2-conjugated O-CMC, O-CMC/Rh2) (106) (see Figure 15) via an esterification reaction. The results showed that 106 was highly porous, and the ester bonds in the structure were pH sensitive. At pH 5.8, the release rate of Rh2 was faster in the early stage, so the release rate of Rh2 could be controlled according to the pH changes at the injury site during inflammatory pain. The conjugate of O-carboxymethyl chitosan enhanced the biological efficacy of Rh2 in vivo by increasing its solubility, regulating its release rate, and prolonging its duration of action in the body. thereby enhancing the biological efficacy of Rh2 in vivo.
Polyethylene glycol (PEG) has the advantages of being easy to modify, biodegradable, biocompatible and having a high drug encapsulation rate, and thus shows great promise in drug delivery. Mathiyalagan et al. [54] combined hydrophilic PEG with hydrophobic Rh1 and Rh2 to synthesize two types of passive targeted delivery ginsenoside derivatives (see Figure 15). Compared with Rh1, PEG-Rh1 (107) has higher antitumor activity in human lung cancer cell lines (A549), while PEG-Rh1 and PEG-Rh2 do not exhibit cytotoxicity in an uninfected murine macrophage cell line (RAW 264.7). Among them, PEG-Rh2 (108) can greatly inhibit nitric oxide production and thus exhibit better anti-inflammatory activity. This indicates that PEG polymers can not only improve the solubility of ginsenosides and reduce cytotoxicity, but also achieve targeted delivery through the enhanced permeability and retention (EPR) effect and different pH conditions.
2.5 Conjugate modification
It has been reported that TPP conjugates have strong mitochondrial targeting ability, and have been used to selectively deliver anticancer drugs, including adriamycin and cisplatin, to the mitochondria of tumor cells [55]. In order to improve the targeting and activity of ginsenoside 25-MeO-PPD, 25-OH-PPD and PD, Ma et al. [56] introduced alkyl chains of different lengths at their C-3 positions, and then conjugated triphenylphosphine (TPP) at the end to synthesize a series of ginsenoside conjugates (see Figure 16). Anti-proliferation studies on cancer cell lines (A549, MCF-7) and normal cells (GES-1) showed that most of the conjugates were more active than the corresponding parent compounds, and exhibited stronger inhibitory effects in cancer cells than in normal cells. Among them, 109 can accumulate in the mitochondria of MCF-7 cells, stimulate the production of reactive oxygen species (ROS), and cause depolarization of the mitochondrial membrane potential, leading to apoptosis. Therefore, 109 exhibits high selectivity and a good antiproliferative effect (IC50 = 0.76 μmol/L) on MCF-7 cells.
2.6 Other modifications
In addition to the five methods mentioned above, structural modifications of ginsenosides also include etherification, alkylation, catalytic hydrogenation, and glycosylation [57-58]. Etherification and alkylation involve the reaction of the hydroxyl group of ginsenosides with haloalkanes under the catalysis of alkalis. Catalytic hydrogenation involves the use of a catalyst to directly hydrogenate the unsaturated group in the monomeric structure of ginsenosides to a saturated group. Glycosylation involves the introduction of corresponding donor groups such as mannosyl, xylosyl and rhamnosyl groups into the hydroxyl group of ginsenosides. For example, Ren et al. [59] introduced sugar donors to the C-20 OH of PPD derivatives through oxidation, reduction, nucleophilic substitution, and other reactions to prepare a series of ginsenoside C-K derivatives with different sugar rings [60-65].
3 Summary
This paper reviews the structural modification methods of ginsenosides in recent years. It mainly uses organic acids, amino acids and inorganic acids to react with the hydroxyl groups at the C-3 and C-20 positions of the aglycone and the primary hydroxyl groups on the sugar chain of ginsenosides to obtain ester derivatives, in order to improve the lipid solubility and bioavailability of ginsenosides. Studies on structure-activity relationships have shown that the activity of ginsenoside derivatives after esterification modification has the following characteristics: unsaturated fatty acids > saturated fatty acids, short-chain fatty acids > long-chain fatty acids. Sulphate modification by introducing polar groups, oxidation modification by reducing unsaturation or adding ionizable groups such as carboxyl groups, heterocyclic modification by increasing the number of hydrogen bonds, and hydrophilic complex modification can all improve the water solubility and bioavailability of ginsenosides to varying degrees, and can significantly improve the biological activity of these derivatives. These modification methods provide an important reference for the study and development and application of ginsenosides.
However, there are currently some deficiencies in the structural modification of ginsenosides: firstly, there are relatively few structural modification methods. Structural modification mainly involves introducing groups that can react with hydroxyl groups, making the sites and types of products of structural modification relatively simple, and leading to insufficient research on the structure-activity relationship. In particular, there is a relative lack of modification of the sugar chain. There are few reports on the replacement of the sugar chain and the splicing of the sugar chain essential for activity with other different types of aglycon or skeleton to enhance activity and broaden the scope of activity. Second, the lack of precision in structural modification results in low activity. Current research on the activity of ginsenoside derivatives mainly focuses on in vitro anti-tumor and anti-oxidant activities, and there is relatively little further research on in vivo activity, with very few compounds entering clinical research. Third, the research on the activity of ginsenoside derivatives is not in-depth enough. There is very limited research on ginsenosides and their derivatives with immunostimulatory activity as vaccine adjuvants. The few studies on ginsenoside adjuvants mainly use crude ginsenoside extracts, and there is a lack of systematic research on the potential application value of ginsenosides and their derivatives as potent immunostimulants in vaccine adjuvants.
Fourth, current structural modifications mainly focus on dammarane-type ginsenosides, with relatively few modifications to oleanolic acid and orcinol-type ginsenosides. Therefore, future structural modifications should improve the accuracy of introducing groups and structures, expand structural modification methods and modification sites, expand the application scope of ginsenoside derivatives, and lay a theoretical foundation for the development of ginsenoside drugs and health foods.
Reference:
[1]HOU M, WANG R, ZHAO S, et al. Ginsenosides in panax genus and their biosynthesis [J]. Acta Pharmaceutica Sinica B, 2021, 11 (7): 1813-1183.
[2]WANG Y H, AI Z Y, ZHANG J D, et al. Research progress on antitumor activity and mechanism of ginsenosides [J]. Science and Technology of Food Industry, 2023, 44(1): 485-491.
[3]PAIk S, SONG G Y, JO E K, et al. Ginsenosides for therapeutically targeting inflammation through modulation of oxidative stress[J]. International Immunopharmacology, 2023, 121: 110461.
[4]LIU F X, LIN Z X, ZHANG H L, et al. Analysis of anti-fatigue mechanism and potential targets of ginseng [J]. China Journal of Chinese Materia Medica, 2019, 44(24): 5479-5487.
[5]ARRING N M, MILLSTINE D, MARKS L A, et al. Ginseng as a treatment for fatigue: A systematic review [J]. JAltern Complement Med. 2018, 24(7):624-633.
[6]FENG S, LI T, WEI X, ZHENG Y, et al. The antioxidant and anti-Fatigue effects of rare ginsenosides and γ-aminobutyric acid in fermented ginseng and germinated brown rice puree. International Journal of Molecular Sciences. 2024, 25(19):10359.
[7]HU Q R, HONG H, ZHANG Z H, et al. Methods on improvements of the poor oral bioavailability of ginsenosides: Pre-processing, structural modification, drug combination, and micro- or nano- delivery system [J]. Journal of Ginseng Research, 2023, 47 (6): 694-705.
[8]FAN W, FAN L, WANG Z, et al. Rare ginsenosides: A unique perspective of ginseng research [J]. Journal of Advanced Research, 2024, https://doi.org/10.1016/j.jare.2024.01.003.
[9]XU W, LYU W, DUAN C, et al. Preparation and bioactivity of the rare ginsenosides Rg3 and Rh2 : An updated review [J]. Fitoterapia, 2023, 167: 105514.
[10]LI S, LI J, ZHAO Y, et al. Supramolecular integration of multifunctional nanomaterial by mannose-decorated azocalixarene with ginsenoside Rb1 for synergistic therapy of rheumatoid arthritis [J]. ACS Nano, 2023, 17 (24): 25468-25482.
[11]YAN H, JIN H, FU Y, et al. Panax ginseng production of rare ginsenosides Rg3 and Rh2 by endophytic bacteria from [J]. Journal of Agriculture and Food Chemistry, 2019, 67 (31): 8493-8499.
[12]XU X, QU W, JIA Z, et al. Effect of cultivation ages on anti-inflammatory activity of a newtype of red ginseng [J]. Biomedicine & Pharmacotherapy, 2021, 136: 111280.
[13]BEDNARCZYK-CWYNAR B, LEŚKÓWA, SZCZUKA I, et al. The effect of oleanolic acid and its four new semisynthetic derivatives on human mewo and a375 melanoma cell lines [J]. Pharmaceuticals, 2023, 16 (5): 746.
[14]DENG X, KE J, ZHENG Y, et al. Α synthesis and bioactivities evaluation of oleanolic acid oxime ester derivatives as -glucosidase and -amylase inhibitors [J]. Journal of Enzyme Inhibition and Medicinal Chemistry, 2022, 37 (1): 451-461.
[15]CAO Y, WANG K, WANG J, et al. Design, synthesis and antibacterial evaluation of ocotillol derivatives with polycyclic nitrogen-containing groups [J]. Future Medicinal Chemistry, 2021, 13 (12): 1025-1039.
[16]LIU J, GAN H, LI T, et al. The metabolites and biotransformation pathways in vivo after oral administration of ocotillol [J]. Biomedical Chromatography, 2020, 34 (8): e4856.
[17]ZHANG D, CAO Y, WANG K, et al. Design, synthesis, and antibacterial evaluation of novel ocotillol derivatives and their synergistic effects with conventional antibiotics [J]. Molecules, 2021, 26 (19): 5969.
[18]TONG Y, SONG X, ZHANG Y, et al. Insight on structural modification, biological activity, structure-activity relationship of PPD-type ginsenoside derivatives [J]. Fitoterapia, 2022, 158: 105135.
[19]GUO H, XING Y, SUN Y, et al. Ginsengenin derivatives synthesized from 20(R)-panaxotriol: Synthesis, characterization, and antitumor activity targeting HIF-1 pathway [J]. Journal of Ginseng Research, 2022, 46 (6): 738-749.
[20]HU Q, HONG H, ZHANG Z, et al. Methods on improvements of the poor oral bioavailability of ginsenosides: Pre-processing, structural modification, drug combination, and micro- or nano- delivery system [J]. Journal of Ginseng Research, 2023, 47 (6): 694-705.
[21]LI J, DAI Y L, ZHENG F, et al. Oral absorption and in vivo biotransformation of ginsenosides [J]. Chinese Journal of Biologicals, 2014, 27(12):1633-1636.
[22]LIANG J, TANG X, WAN S, et al. Structure modification of ginsenoside Rh2 and cytostatic activity on cancer cells [J]. ACS Omega, 2023, 8 (19): 17245-17253.
[23]LI K K, YAN X M, LI Z N, et al. Synthesis and antitumor activity of three novel ginsenoside M1 derivatives with 3′-ester modifications [J]. Bioorganic Chemistry, 2019, 90: 103601.
[24]HUANG Y, LI H M, ZHANG Y X, et al. Synthesis and biological evaluation of ginsenoside compound K derivatives as a novel class of lxrα activator [J]. Molecules, 2017, 22 (7): 1232.
[25]WANG R, LI M, LIU M, et al. Characterization of pickering emulsion by SCFAs-modified debranched starch and a potent for delivering encapsulated bioactive compound [J]. International Journal of Biological Macromolecules, 2023, 231: 123164.
[26]SUNG KEE R, TIMONTHY J K, KAZUTOSHI F, et al. Effect of an oral astaxanthin prodrug (CDX-085) on lipoprotein levels and progression of atherosclerosis in ldlr -/- and apoe -/- mice [J]. Atherosclerosis, 2012, 222.
[27]CHRISTOPHER T C, UDAYANATH A, CHRISTOPHER A W, et al. Targeting pro-invasive oncogenes with short chain fatty acid-hexosamine analogues inhibits the mobility of metastatic mda-mb-231 breast cancer cells [J]. Journal of Medicinal Chemistry, 2008, 51:8135-8147.
[28]ZHANG J, TONG Y, LU X, et al. A derivant of ginsenoside C-K and its inhibitory effect on hepatocellular carcinoma [J]. Life Sciences, 2022, 304: 120698.
[29]XIAO S, LIN Z, WANG X, et al. Synthesis and cytotoxicity evaluation of panaxadiol derivatives [J]. Chemistry & Biodiversity, 2020, 17 (1): e1900516.
[30]YUAN W,GUO J, WANG X,et al.Non-protein amino acid derivatives of 25-methoxylprotopanaxadiol/25-hydroxyprotopanaxadioland their anti-tumor activity evaluation [J]. Steroids, 2018, 129: 1-8.
[31]LIN L, ZHAO Y, WANG P, et al. Amino acid derivatives of ginsenoside AD-2 induce HepG2 cell apoptosis by affecting the cytoskeleton [J]. Molecules, 2023, 28 (21): 7400.
[32]MIYAMOTO T, TOGAWA K, HIGUCHI R, et al. Six newly identified biologically active triterpenoid glycoside sulfates from the sea cucumber cucurnaria echinata [J]. Liebigs Annalen der Chemie, 1989 1990 (5):453-460.
[33]GUO Z, WANG L, HAQ S, et al. In-vitro evaluation of immunomodulatory activity of sulphation-modified total ginsenosides derivative-3 [J]. Frontiers in Veterinary Science, 2023, 10: 1068315.
[34]FU B, BI W, HE C, et al. Sulfated derivatives of 20(S)-ginsenoside Rh2 and their inhibitory effects on LPS-induced inflammatory cytokines and mediators [J]. Fitoterapia, 2013, 84: 303-307.
[35]BI W, FU B, SHEN H, et al. Sulfated derivative of 20(S)-ginsenoside Rh2 inhibits inflammatory cytokines through mapks and nf-kappa b pathways in LPS-induced RAW264.7 macrophages [J]. Inflammation, 2012, 35: 1659-1668.
[36]YIP, BI W, SHEN H, et al. Inhibitory effects of sulfated 20(S)-ginsenoside Rh2 on the release of pro-inflammatory mediators in LPS-induced RAW 264.7 cells [J]. European Journal of Pharmacology, 2013, 712: 60-66.
[37]ZHOU W X, YANG N, ZHAO Y Q. Advances in studies on improvement of water solubility of ginsenoside[J]. Drug Evaluation Research, 2016, 39(2): 322-327.
[38]WONG V, DONG H, LIANG X, et al. Rh2E2, a novel metabolic suppressor, specifically inhibits energy-based metabolism of tumor cells [J]. OncoTargets and Therapy, 2016, 7 (9): 9907-9924.
[39]SUN Y X, FANG X J, GAO M, et al. Synthesis and structure−activity relationship of pyxinol derivatives as novel anti-inflammatory agents [J]. ACS Medicinal Chemistry Letter, 2020, 11, 457-463.
[40]WANG Y, MI X, DU Y, et al. Design, synthesis, and anti-inflammatory activities of 12-dehydropyxinol derivatives [J]. Molecules, 2023, 28 (3): 1307.
[41]YANG G, Mi X, WANG Y, et al. Fusion of michael-acceptors enhances the anti-inflammatory activity of ginsenosides as potential modulators of the NLRP3 signaling pathway [J]. Bioorganic Chemistry, 2023, 134:106467.
[42]ZHANG Y M, YUAN W H, WANG X D, et al. Synthesis, characterization and cytotoxic activity evaluation of ginsengdiol oxidation and nitrogen hybrid derivatives [J]. MedChemComm, 2018, 9(11): 1910-1919.
[43]XIAO S, WANG X, XU L, et al. Novel ginsenoside derivatives have shown their effects on PC-3 cells by inducing G1-phase arrest and reactive oxygen species-mediate cell apoptosis [J]. Bioorganic Chemistry, 2021, 112: 104864.
[44]YANG G Q, LIU S, ZHANG C, et al. Discovery of pyxinol amide derivatives bearing amino acid residues as nonsubstrate allosteric inhibitors of p-glycoprotein-mediated multidrug resistance [J]. Journal of Medicinal Chemistry, 2023, 66 (13): 8628-8642.
[45]MA L, MIAO D, LEE J, et al. Synthesis and biological evaluation of heterocyclic ring-fused dammarane-type ginsenoside derivatives as potential anti-tumor agents [J]. Bioorganic Chemistry, 2021, 116: 105365.
[46]KARROUCHI K, RADI S, RAMLI Y, et al. Synthesis and pharmacological activities of pyrazole derivatives: A review [J]. Molecules, 2018, 23(1):134.
[47]WANG J, WANG D B, SUI L L, et al. Natural products-isoxazole hybrids: A review of developments in medicinal chemistry [J]. Arabian Journal of Chemistry, 2024, 17(6):105794.
[48]DAI R, WEI X, LI T, et al. Synthesis and antitumor activity of panaxadiol pyrazole and isooxazole derivatives [J]. Chemistry & Biodiversity, 2023, 20 (8): e202300507.
[49]HOU W, DAI W, HUANG H, et al. Pharmacological activity and mechanism of pyrazines [J]. European Journal of Medicinal Chemistry, 2023, 258:115544.
[50]RASHID H U, MARTINES M A U, DUARTE A P, et al. Research developments in the syntheses, anti-inflammatory activities and structure-activity relationships of pyrimidines [J]. RSC Advance. 2021, 11(11): 6060-6098.
[51]WANG S, ZHANG J, ZHANG J, et al. Synthesis and biological evaluation of heterocyclic ring-fused 20(s)-protopanaxadiol derivatives as potent antiosteoporosis agents [J]. Journal of Medicinal Chemistry, 2023, 66 (17): 11965-11984.
[52]YANG K, YANG Z, Yu G, et al. Polyprodrug nanomedicines: An emerging paradigm for cancer therapy [J]. Advanced Materials, 2022, 34 (6): e2107434.
[53]LU H, CEN J, REN Y, et al. Evaluation of the anti-inflammatory pain effect of ginsenoside-conjugated o-carboxymethyl chitosan particles [J]. Polymers, 2023, 15 (19): 4011.
[54]MATHIYALAGAN R, WANG C, KIM Y, et al. Preparation of polyethylene glycol-ginsenoside Rh1 and Rh2 conjugates and their efficacy against lung cancer and inflammation [J]. Molecules, 2019, 24 (23): 4367.
[55]BATHEJA S, GUPTA S, TEJAVATH K K, et al. TPP-based conjugates: potential targeting ligands [J]. Drug Discovery Today. 2024, 29(6):103983.
[56]MA L, WANG X, LI W,et al.Rational design,synthesis and biological evaluation of triphenylphosphonium-ginsenoside conjugates as mitochondria-targeting anti-cancer agents [J]. Bioorganic Chemistry, 2020, 103: 104150.
[57]ZHANG H R, YE AQ, ZHANG Y W, et al. Research progress on ginsenoside derivatization and its biological activities [J]. Chinese Traditional and Herbal Drugs, 2022, 53(14): 4554-4567.
[58]YUAN S Z, WANG B, Zhou X, et al. Research progress on the biotransformation of rare ginsenosides [J].Science and Technology of Food Industry, 2023, 44(12): 480-489.
[59]RRN S, LIU R, WANG Y, et al. Synthesis and biological evaluation of ginsenoside compound K analogues as a novel class of anti-asthmatic agents [J]. Bioorganic & Medicinal Chemistry Letters, 2019, 29 (1): 51-55.
[60]REN G, LV W, DING Y, et al. Ginseng saponin metabolite 20(S)-protopanaxadiol relieves pulmonary fibrosis by multiple-targets signaling pathways [J]. Journal of Ginseng Research, 2023, 47 (4): 543-551.
[61]ZHANG H, ZHANG L, YANG C, et al. Prevention effect of protopanaxadiol-type saponins saponins and protopanaxatriol-type saponins on myelosuppression mice induced by cyclophosphamide [J]. Frontiers in Pharmacology, 2022, 13: 845034.
[62]KIM A, PARK SM, KIM NS, et al. Ginsenoside Rc, an active component of panax ginseng, alleviates oxidative stress-induced muscle atrophy via improvement of mitochondrial biogenesis [J]. Antioxidants, 2023, 12 (8).
[63]LI S, LI JJ, ZHAO YY, et al. Supramolecular integration of multifunctional nanomaterial by mannose-decorated azocalixarene with ginsenoside Rb1 for synergistic therapy of rheumatoid arthritis [J]. ACS Nano, 2023, 17 (24): 25468-25482.
[64]LEE H, KONG G, TRAN Q, et al. Relationship between ginsenoside Rg3 and metabolic syndrome [J]. Frontiers in Pharmacology, 2020, 11: 130
[65]CHEN Y Y, LIU Q P, AN P, et al. Ginsenoside Rd: A promising natural neuroprotective agent [J].Phytomedicine, 2022, 95: 153883.







